Recent Topics
Last update on May 25,
2011
See also our publication
list.
Charge Transfer and Molecular Orientation of
Tetrafluorotetracyanoquinodimethane on a Hydrogen-Terminated Si(111)
Surface Prepared by a Wet Chemical Method [Masayuki
Furuhashi and Jun Yoshinobu, J. Phys. Chem. Lett. 1(2010)
pp.2917-2921]
We investigated the chemical state and molecular orientation of
2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4-TCNQ)
adsorbed on a hydrogen-terminated Si(111)(1x1) surface using
transmission infrared (IR) spectroscopy. We deposited F4-TCNQ
molecules on H-Si(111) by a wet chemical method. Similar to
evaporated F4-TCNQ molecules on various substrates in vacuum, we
observed anionized F4-TCNQ on the H-Si(111) substrate. The incident
angle dependence of the IR spectra reveals that this F4-TCNQanion
lies flatly on the surface. On the other hand, minority neutral
F4-TCNQ species assume random orientation, judging from the
comparison between s- and p-polarized IR spectra. We conclude that
the first layer on the H-Si surface is a flat-lying anion species and
the upper layers consist of randomly oriented neutral molecules.
Infrared spectroscopy of the organic monolayer
sandwiched between a Hg electrode and a Si substrate [Masayuki
Furuhashi and Jun Yoshinobu, Rev. Sci. Instrum. 81, 053103 (2010);
doi:10.1063/1.3422256 (6 pages) ]
We have successfully observed the vibrational spectra of organic
monolayers sandwiched between a liquid Hg electrode and a Si
substrate by means of a newly developed reflection absorption (RA)
device(Fig. 1). The vibrational spectra of organic monolayers between
two electrodes can be observed under a certain bias voltage. The
monolayers were fabricated by the reaction of hydrogen-terminated
Si(111) with 1-octadecene. A metal/insulator/semiconductor structure
was prepared using liquid Hg as a metal electrode and the organic
monolayer as an insulator. Infrared (IR) light entered from the Si
substrate side with an incident angle of 75°. The reflected IR
light from the metallic Hg was detected by a
mercury-cadmium-telluride detector. We obtained RA spectra using a
bare H&endash;Si(111) substrate as a reference. The absorbance of the
RA spectrum (Fig. 2b) was comparable with that of the transmission
spectrum (Fig.2a) for the octadecyl-terminated Si(111) without Hg.
The C&endash;H stretching modes in the CH2
group show blueshifts, and the C&endash;H antisymmetric stretching
modes in the CH3 are broadened in comparison
with the transmission spectrum. Under a certain bias voltage, we
observed changes in band shape. We concluded that the variation was
due to the temperature increase by resistive heating of the
substrate.
Fig.1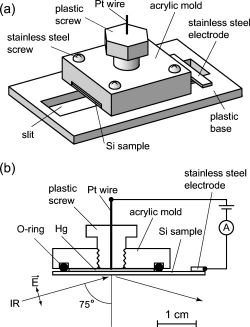 Fig.2
Fig.2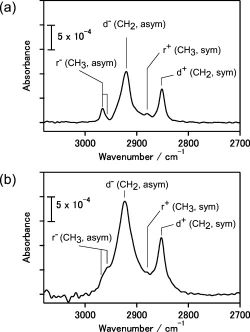
Low-temperature observation of the softened C-H stretching vibrations
of cyclohexane on Rh(111)
[Takanori Koitaya, Atsushi Beniya, Kozo Mukai, Shinya Yoshimoto, and
Jun Yoshinobu, Phys. Rev. B 80, 193409 (2009) DOI:
10.1103/PhysRevB.80.193409 ].
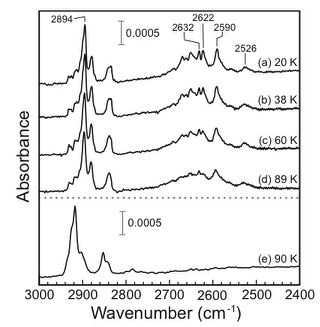
The C-H stretching vibrations of cyclohexane on
Rh(111) were investigated using infrared reflection absorption
spectroscopy between 20 and 89 K. At 20 K, the softened C-H
stretching band consists of several sharp peaks, ranging from 2500 to
2700ÝcmÄ|1. The wide-range distribution of the softened
C-H stretching peaks results from inhomogeneity of adsorption
environments. With increasing the substrate temperature, each
softened C-H stretching peak becomes significantly broadened, but the
normal C-H stretching peaks are little changed. These results
indicate that the local interaction between the softened C-H species
and the Rh(111) surface is sensitive to a thermally excited
low-energy mode. The temperature-dependent broadening of a soft mode
at low temperature is analyzed using a vibrational dephasing model,
where the softened C-H stretching mode is anharmonically coupled with
a thermally excited frustrated translation mode.
The growth process of first water layer and crystalline ice on the
Rh(111) surface [Atsushi
Beniya, Yuji Sakaguchi, Tetsuya Narushima, Kozo Mukai, Yoshiyuki
Yamashita, Shinya Yoshimoto, and Jun Yoshinobu; J. Chem. Phys. 130,
034706 (2009); doi:10.1063/1.3060952 (10 pages)
The adsorption states and growth process of the first layer and
multilayer of water (D2O) on Rh(111) above 135ÝK were
investigated using infrared reflection absorption spectroscopy
(IRAS), temperature programed desorption, spot-profile-analysis
low-energy electron diffraction, and scanning tunneling microscopy
(STM). At the initial stage, water molecules form commensurate
(�3Ý�3)R30° islands, whose size is limited for several
hexagonal units; the average diameter is Ý 2.5Ýnm. This
two-dimensional (2D) island includes D-down species, and free OD
species exist at the island edge. With increasing coverage, the D-up
species starts to appear in IRAS. At higher coverages, the 2D islands
are connected in STM images (Fig.1). By the titration of Xe
adsorption we estimated that the D-down domain occupies about 55% on
Rh(111) at the saturation coverage. Further adsorption of water
molecules forms three-dimensional ice crystallites on the first water
layer; thus, the growth mode of crystalline water layers on Rh(111)
is a Stranski&endash;Krastanov type. We have found that an ice
crystallite starts to grow on D-down domains and the D-down species
do not reorient upon the formation of a crystalline ice.
 Fig. 1
Fig. 1
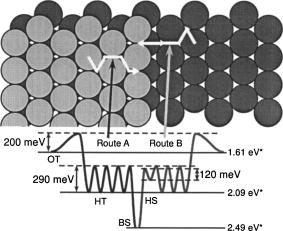
Microscopic diffusion processes of NO on the
Pt(997) surface [Noriyuki
Tsukahara, Kozo Mukai, Yoshiyuku Yamashita, and Jun Yoshinobu,
J.
Chem. Phys. 128(2008)054701]
The microscopic diffusion processes of NO molecules on Pt(997) at
low coverage were investigated using time-resolved infrared
reflection absorption spectroscopy (TR-IRAS). When NO molecules
adsorb on Pt(997) at low temperature, each molecule transiently
migrates on the surface from the first impact point to a possible
adsorption site. At 11 K, the molecules are trapped at
four adsorption sites on Pt(997): the on-top sites on the (111)
terrace (OT), the hollow sites on the (111) terrace (HT), the bridge
sites at the step (BS) and the hollow sites at the step downstream
(HS). Based on the initial population ratio for these sites, the mean
lateral displacement by transient migration is estimated to be
4.1 Å. By heating the surface to 45 K,
the HS species migrate up to the BS sites; the migration barrier is
roughly estimated to be 120 meV. In the temperature range
from 70 to 77 K, TR-IRAS
measurements were carried out to observe the site change of OT
species to the adjacent HT sites at isothermal conditions; the
activation barrier and the preexponential factor are estimated to be
200 meV and
2.0x1011 s-11, respectively. In the
temperature range from 100 to 110 K,
the HT species migrate across the terrace and finally reach the BS
sites. The activation barrier between the HT sites and the
preexponential factor are estimated to be 290 meV and
6.5x1011 s-11, respectively, from
the TR-IRAS data together with kinetic Monte Carlo simulations. On
the whole, the quantitative microscopic picture of NO migration on
Pt(997) has been established.
Regioselective Cycloaddition Reaction of
Alkene Molecules with the Asymmetric Dimer on
Si(100)c(4x2) [K. Oguchi et al., J.
Am. Chem. Soc. 129(2007)
1242-1245.]
We investigated the adsorption states of 2-methylpropene and
propene on Si(100)c(4x2) using low-temperature scanning tunneling
microscopy. We have found that regioselective cycloaddition reactions
(di-sigma bond formation) occur between the asymmetric alkene
molecules and the asymmetric dimers on Si(100)c(4x2).
First-principles calculations have elucidated that the
regioselectivity is closely related to the structures of precursor
species and these precursor species have carbocation-like features.
Thus, we conclude that Markovnikov's rule is applicable for the
cycloaddition of asymmetric alkene with the asymmetric dimer on
Si(100)c(4x2).

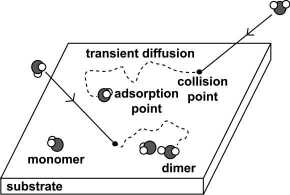
Transient diffusion and cluster formation of water
molecules on Rh(111) at 20 K [ Atsushi Beniya, Kozo
Mukai, Yoshiyuku Yamashita and Jun Yoshinobu, J.
Chem. Phys. 126 (2007)141102]
We investigated the initial stage of water adsorption on Rh(111)
at 20 K using infrared reflection absorption spectroscopy (IRAS). In
this low coverage region, isolated water molecules and small water
clusters are observed. Since thermal diffusion is suppressed at 20 K,
the formation of water clusters at low coverage is controlled by both
coverage and transient diffusion on the surface. Within a simple
random walk model as transient diffusion and clustering process, we
estimate the mean lateral displacement from the first impact point to
the final adsorption site to be 0.76 nm.
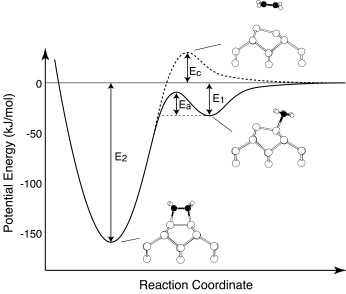
Precursor mediated cycloaddition reaction
of ethylene to the Si(100)c(4x2) surface [M.Nagao et al., JACS
126(2004) 9922-9923]
We report the direct observation of a precursor state for the
cycloaddition reaction (the di-s bond
formation) of ethylene on Si(100)c(4x2) using high-resolution
electron energy loss spectroscopy at low temperature, and the
meta-stable precursor state is identified as a weakly bonded p -complex type. The activation energy from the
p-complex precursor to the di- s bonded species is experimentally estimated to
be 0.2 eV. First-principles calculations support the p-complex precursor mediated cycloaddition
reaction of ethylene on Si(100)c(4x2).
Low-Energy Electron-Stimulated Chemical
Reactions of CO in Water Ice [S.
Yamamoto et al., Chem. Phys. Lett. 388(2004) 284-288]
We investigated low-energy electron-stimulated chemical reactions
between CO and water molecules in low-temperature ice using infrared
reflection absorption spectroscopy. Carbon dioxide, the formyl
radical, formaldehyde, and methanol were produced by electron
irradiation of the water/CO/water layered ice.The electron energy
threshold and temperature dependence for the chemical reactions were
investigated to elucidate the reaction mechanisms.
CO in water ice +
e- -> CO2,
H2CO, CH3 OH
Selective functionalization of the Si(100) surface by converting the
adsorption linkage of a bifunctional organic molecule [Md. Zakir Hossain et al., Chem. Phys. Lett.
388(2004) 27-30]
Selective functionalization by converting the adsorption linkage of
bi-functional organic molecule, 1-dimethylamino-2-propyne (DMAP), on
Si(100) has been studied using high-resolution electron energy loss
spectroscopy (HREELS) and scanning tunneling microscopy (STM). The
HREELS and STM results clearly indicate that DMAP adsorbs at the down
dimer atom of Si(100)c(4x2) through an N-Si dative bond leaving the
acetylene unit of DMAP free at low temperature (65 ~ 90 K). The
adsorption linkage is converted from the N-Si dative bond to the Si-C
di- s bonds by annealing the adsorbed
surface to 300 K, and thus the tertiary amino group becomes free.
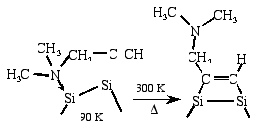
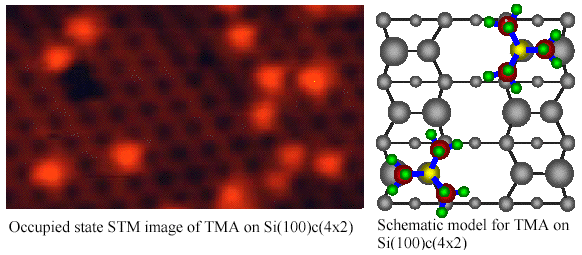
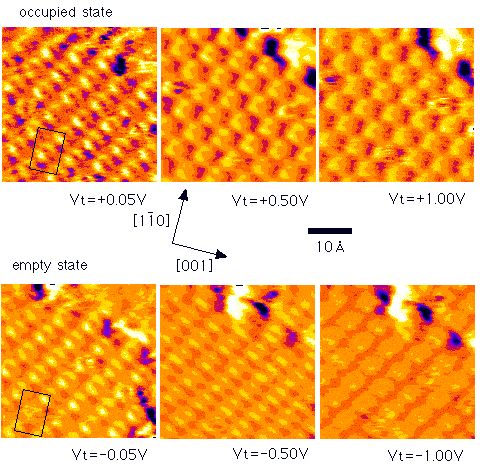
Purely site-specific
chemisorption and conformation of trimethylamine on
Si(100)c(4x2)[Md. Zakir Hossain et al., JACS 125(2003)
9252-9253]
We have investigated the
adsorption of trimethylamine (TMA) on Si(100)c(4x2) using scanning
tunneling microscopy (STM) at 80 K. The adsorbed TMA appears as a
triangle shaped bright protrusion in the occupied state STM image.
The triangle shaped protrusion is ascribed to three methyl groups in
the adsorbed TMA. The center of the protrusion is located on the down
atom site, which indicates that the adsorption of TMA occurs only on
the down dimer atom. Thus, TMA adsorption on Si(100)c(4x2) is found
to be purely site-specific on the down dimer atom and can be
categorized in Lewis acid-base reaction.
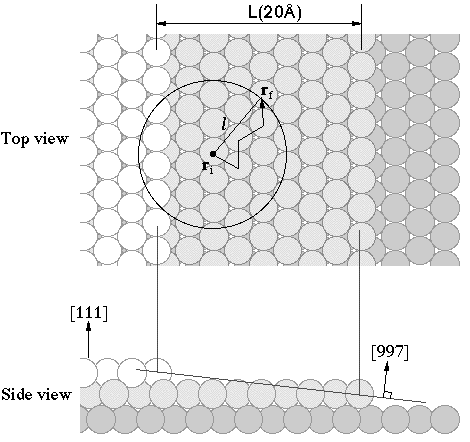
Lateral displacement by transient mobility in chemisorption of CO on
Pt(997)
(Yoshinobu et
al., Phys. Rev. Lett. 90(2003)248301 )
We investigated adsorbed states
of CO on Pt(997) at 11 K using infrared reflection absorption
spectroscopy. At 11 K, thermal migration is suppressed and thus the
initial chemisorption at terrace sites and step sites is controlled
by the transient mobility of the adsorbing molecule. The initial
occupation ratio between atop CO on the terrace and atop CO at the
step is directly determined to be 3.6:1.With a simple isotropic
migration model, we estimated the mean lateral displacement from the
first impact point to the initial chemisorption site to be 0.68nm .We also discuss the
origin of transient mobility of CO on metal surfaces.
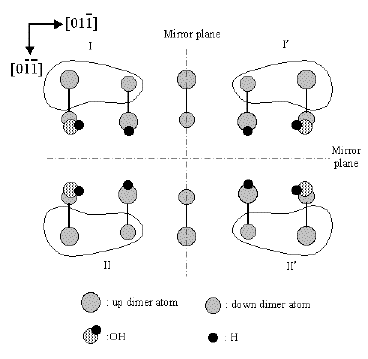
Elucidation of c-type defect on
Si(100)
(Md.Zakir Hossain et al., Phys. Rev. B. 67(2003)153307. )
The origin and atomic structure of C-defect on Si(100) have been
unambiguously identified. Two pairs of enantiomorphic protrusions of
C-defect (U,U',D,D') have been observed by low temperature scanning
tunnelling microscopy (STM). These are attributed to the dissociative
adsorption of single water molecule on two adjacent dimers. Two
unreacted dangling bonds on these dimers have different electronic
states which are visualized in unoccupied state STM images.

Si 2p photoelectron spectrum of
Si(100)c(4x2) at 30K (S. Machida et
al., Surf. Sci. 532/535 (2003) 716-720)
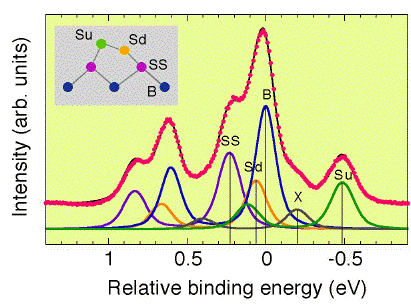
This spectrum was measured with hn=129eV using a Phoibos100
analyzer at KEK-PF BL16B. The overall instrumental resolution was
~40meV, so that the width of each component can be estimated as a
function of temperature.
Direct evidence for asymmetric dimer of Si(100) surface at low
temperatures by means of high resolution Si 2p photoelectron
spectroscopy (Y. Yamashita et al., Jpn. J. Appl. Phys
41(2002)L272-L274.)
Surface atoms on Si(100) are reconstructed to form a (2x1) dimer
row structure. Many experimental and theoretical studies have
supported the buckled (asymmetric) dimer with c(4x2) phase as a
ground state, although some quantum chemical studies including
electron correlation have supported a symmetric dimer as the most
stable state. Recently, several low temperature STM studies have
reported symmetric dimer images. With decreasing the temperature from
80 K to 20 K, the area of apparent symmetric dimer increases. The
origin of symmetric images is a controversial issue (dynamic or
static). In this study, using high resolution Si 2p photoelectron
spectroscopy, we have investigated the electronic states of Si(100)
at 140 K, 100 K and 55 K (Fig.1). By careful analysis of surface core
level shift and area intensity of each component in the spectra, the
number of asymmetric dimer atoms does not change in this temperature
range. Thus, we conclude that the dimer on Si(100) is asymmetric down
to 55 K, and the origin of symmetric images in STM may be due to
dynamic effects.
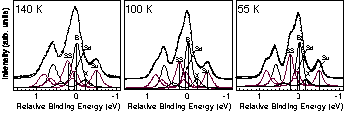
Fig.1 Si 2p PES spectra at low temperature. (hn=129eV, normal emission)
High-resolution core-level photoelectron spectroscopy of organic and
inorganic molecules on Si(100)
[ISSP Yoshinobu laboratory: since December
1999]
Chemical bond between adsorbed unsaturated
hydrocarbon molecules and the Si(100)(2x1) surface has been
investigated by means of HRPES at PF
BL-16B. This
undulater beam line has a 24-m High-resolution Spherical Grating
Monochromator (H-SGM), which covers the photon energy range from 40
eV to 550 eV. We have carried in our UHV chamber including a LEED
optics (Varian) and an old electron analyzer (VSW HA100) and a new
SPECS Phoibos100.
Microscopic behavior of atoms and molecules on surfaces by using a
combined low temperature STM and IRAS system
[ISSP Yoshinobu laboratory: since August
1999]
In order to elucidate microscopic behavior and nano structures
of atoms/molecules on solid surfaces, we are constructing a new
apparatus which includes low temperature STM and IRAS. At the present
moment, the IRAS system shows a satisfactory good S/N ratio
(<3x10-5 abs: 1000scans, 4cm-1).

Towards the fabrication of hybrid system with organic molecules on
Si(100)
[ISSP Yoshinobu laboratory &
IMS-UVSOR]
In the cases of cyclopentene and cyclohexene, the dangling bond
state just below the Fermi level in valence PES spectra has decreased
in intensity with increasing the coverage and at the saturation the
dangling bond peak disappears almost completely. On the other hand,
the HOMO derived states which are clearly observed for the multilayer
of molecules at 90K have been significantly attenuated at the
original position. Thus, we conclude that the p
bond of cyclopentene (and cyclohexene) interacts with the Si
dangling bonds. According to the Si 2p spectra, it seems that the SiC
bonds are formed between the Si surface and the molecule. This is
also supported from the information about the C 1s peak in XPS
spectra taken at ISSP.
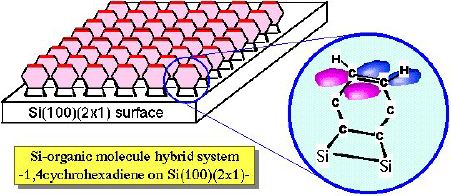
1,4-cyclohexadiene has two C=C double bonds. There are two possible
adsorption structures on Si(100)(2x1); (1) two p bonds interact with Si dangling bonds to form
four SiC sigma bonds, and (2) one p bond
reacts with one Si dimer to make di-sigma bond and another C=C bond
is still intact. Preliminary results from PES in UVSOR-BL5A and XPS
in ISSP suggest that the case (2) is plausible. In this case,
additional molecular development/modification is possible using
organic chemical reactions with 1,4-cyclohexadiene/Si(100). However,
further experiments are needed to confirm the adsorption structure
and realize this idea.
A new photoelectron spectroscopy apparatus for solid surfaces at low
temperature
[ISSP Yoshinobu laboratory]
In order to elucidate electronic properties of adsorbed
atoms/molecules and surfaces, a new UHV system including a
hemispherical electron analyzer (VG CLAM4), X-ray sources (VG XR3), a
UV source (VG UVL-Hi) with a polarizer, LEED (VSI), QMS (Balzers
Prisma) and the home-made gas dosing system (a continuous doser with
a variable temperature tungsten tube and a MCP doser with a pulse
valve) is constructed. The sample is cooled down to <30 K using a
closed cycle He refrigerator, and can be rotated around two axis.

(From left: Ms. Hamaguchi, Dr. Yamashita and Mr. Machida)
Internal structure of STM images in the Pd(110)c(4x2)benzene
[J. Yoshinobu et al., Phys. Rev. Lett.,
79 3942-3945
(1997)]
Evidence for a molecule-substrate hybridized state near the Fermi
level (EF) is presented for Pd(110)c(4x2)-benzene.
Observed images of adsorbed benzene near EF by STM consist
of two elongated protrusions separated by a single nodal depression
with C2 symmetry. The existence of a benzene derived state
near EF is also observed by metastable atom electron
spectroscopy, and it is assigned to the antibonding states between
the 1e1g molecular orbital of benzene and the Pd 4d
orbitals by ab initio molecular orbital calculations.
Determination of adsorption potential energy surface of CO on Ni(100)
[T. Moriwaki, R. Kishi, J. Yoshinobu and
M. Kawai, submitted to J. Chem. Phys.]
The microscopic surface diffusion of CO molecules on Ni(100) has
been studied with infrared reflection absorption spectroscopy
(IRAS). We have observed low temperature kinetics approaching
equilibrium between isolated adsorbed CO and c(2x2)-CO domains at a
low coverage. The upper limit of the diffusion barrier Ed
= 60 ± 7 meV and the preexponential factor A =
106.8±1.1 s-1 were estimated. While the
adsorption energy (Ead) of CO on Ni(100) is approximately
1.3 eV, the diffusion barrier is less than 5% of Ead. It
is shown that the barrier is consistent with the postulated potential
energy surface [J. Yoshinobu et al., Phys. Rev.B49,16670-16677
(1994)] derived from the frustrated translation modes and the binding
energy difference with a harmonic parabola assumption, and also with
the recent microscopic diffusion barrier obtained by quasielastic He
atom scattering (QHAS) measurements.
NiCOpotential.JPG
Thermal excitation of oxygen species as a
trigger for CO oxidation on Pt(111)
[ J. Yoshinobu and M. Kawai, J. Chem. Phys. 103 (1995)
3220-3229]
Thermal excitation of adsorbed oxygen species is found to
initiate the CO oxidation on Pt(111). We have prepared three
different coadsorption systems to study the reactivity of different
oxygen species: (1) CO on the O2 preadsorbed Pt(111)
surface, (2) CO on the nearly perfect Pt(111) p(2x2)-O surface and
(3) CO on the disordered atomic oxygen-preadsorbed Pt(111) surface.
Four CO2 desorption peaks (a-CO2 at 125 K, b3-CO2 at ~225 K, b2-CO2 at ~260 K and b1-CO2 at 320 K) are
observed. The desorption temperatures of CO2 strongly
depend on the adsorbed states of oxygen species. We have shown that
the a-CO2 state, b2,3-CO2states and b1 - CO2 state are
correlated with adsorbed O2, disordered oxygen atoms and
p(2x2) oxygen atoms, respectively. The difference in CO2
desorption temperature is related to thermal excitation of each
oxygen species, which is derived from the structural information of
coadsorbed states during thermal evolution by means of LEED and IRAS.
This proposed mechanism can explain most experimental data of
CO2 formation dynamics on Pt(111), and is supported by a
recent first principle calculation [A.Alavi et al., Phys. Rev. Lett.
80, 3650 (1998)].
Go to Homepage of Yoshinobu
Laboratory
{kind=link}